Negli ultimi cinquant'anni, la comprensione e la classificazione dei disturbi del neurosviluppo hanno subito trasformazioni significative. Particolare attenzione è stata posta sui disturbi dello spettro autistico (ASD) e sulla sindrome di Rett (RTT), condizioni che, pur presentando alcune sovrapposizioni sintomatologiche, sono distinte e richiedono approcci diagnostici e terapeutici specifici. L'evoluzione dei manuali diagnostici, come il DSM-5 e l'ICD-11, ha ridefinito i criteri e le nomenclature, offrendo una visione più sfumata e precisa di queste complesse condizioni neurologiche. Questo articolo si propone di esplorare le modifiche terminologiche e diagnostiche, approfondire le caratteristiche cliniche, le eziologie, le sfide diagnostiche e le attuali prospettive terapeutiche, con un focus particolare sulla sindrome di Rett e il suo rapporto con i disturbi dello spettro autistico.
Evoluzione delle Classificazioni Diagnostiche: Dal DSM-IV-TR al DSM-5 e ICD-11
Le modifiche più significative nella terminologia diagnostica dei disturbi dello spettro autistico sono emerse con la pubblicazione del DSM-5 nel 2013 e dell'ICD-11. Il DSM-5 ha apportato cambiamenti sostanziali rispetto al DSM-IV-TR in termini di denominazione, classificazione, criteri diagnostici, livelli di gravità e prevalenza.
Una delle modifiche più rilevanti introdotte dal DSM-5 è l'abbandono della classificazione in specifici sottotipi di disturbo autistico presenti nel DSM-IV-TR. Ora, chi manifesta marcati deficit nella comunicazione sociale ma non presenta i comportamenti ristretti e ripetitivi tipici dei disturbi dello spettro autistico viene valutato per la diagnosi di Disturbo della Comunicazione Sociale (pragmatica), piuttosto che per un disturbo dello spettro autistico.
Cruciale nel DSM-5 è l'introduzione della specificazione dei livelli di gravità dei sintomi. Viene allegata una tabella che distingue tre diversi livelli di gravità, valutati sulla base delle necessità di supporto, sia per quanto riguarda la "comunicazione sociale" sia per i "comportamenti ristretti e ripetitivi". Questo approccio abbandona la precedente distinzione psicometrica basata sulle deviazioni standard del QI, focalizzandosi invece sul fabbisogno di assistenza.
Con l'ICD-11, si assiste a un ulteriore superamento della vecchia distinzione in vari tipi di autismo. Viene proposta una nuova classificazione che enfatizza la possibile comorbilità con disturbi del linguaggio e disturbi dello sviluppo intellettivo. Sebbene i nuovi codici dell'ICD-11 diventeranno validi a livello pubblico solo con l'adozione da parte dei sistemi sanitari nazionali, questa distinzione solleva importanti considerazioni, specialmente riguardo al concetto di "alto funzionamento". La differenza tra "alto" e "basso funzionamento" viene ora strettamente legata alla presenza o assenza di disabilità intellettiva. In sintesi, si parla di "basso funzionamento" quando il QI totale è inferiore a 70. Pertanto, la dicitura "autismo ad alto funzionamento" necessita di una verifica del QI totale per una corretta interpretazione.
La Sindrome di Rett: Una Patologia Genetica Distinta
La sindrome di Rett (RTT) è una rara patologia neurologica dello sviluppo che colpisce prevalentemente il sesso femminile. Si tratta di una malattia congenita che interessa il sistema nervoso centrale ed è una delle cause più diffuse di deficit cognitivo grave o gravissimo. La sua manifestazione tipica avviene dopo i primi 6-18 mesi di vita, con una perdita progressiva della motricità, delle capacità manuali e dell'interesse all'interazione sociale. L'incidenza stimata varia, ma si aggira intorno a 1 su 9.000 tra le ragazze di 12 anni e 1 su 30.000 nella popolazione generale. Alcune stime indicano 1 bambina su 10.000, rendendola la seconda causa mondiale di disabilità intellettiva femminile.
A differenza dei disturbi dello spettro autistico, la sindrome di Rett è una malattia genetica causata principalmente da mutazioni nel gene MECP2, localizzato sul cromosoma X (Xq28). Questo gene è cruciale per lo sviluppo del cervello, in quanto codifica per la proteina MeCP2, coinvolta nella regolazione dell'espressione genica. Le mutazioni in MECP2, in particolare quelle "loss-of-function" (>95% dei casi classici), alterano questa funzione, portando a un ampio spettro di manifestazioni cliniche. Sebbene la maggior parte dei casi siano mutazioni spontanee, una piccola percentuale (<1%) può essere ereditata.
Le bambine affette da sindrome di Rett presentano tipicamente un periodo iniziale di sviluppo apparentemente normale nei primi sei mesi di vita, durante il quale raggiungono le tappe di crescita previste per la loro età. Successivamente, tuttavia, si osserva una regressione con disfunzioni dell'andatura, perdita delle abilità manuali acquisite e comparsa di movimenti stereotipati e ripetitivi delle mani (come strizzare, battere, sfregare, o portare le mani alla bocca). Possono verificarsi anche disordini respiratori (apnea, iperventilazione), difficoltà nella deambulazione, perdita del linguaggio parlato e disabilità intellettiva.
Sintomatologia e Criteri Diagnostici della Sindrome di Rett
La sindrome di Rett è caratterizzata da una notevole eterogeneità clinica, dovuta alle centinaia di mutazioni che possono interessare il gene MECP2. Questa variabilità genetica si traduce in un profilo patologico non perfettamente sovrapponibile tra i pazienti, costituendo una sfida sia in chiave diagnostica che nello sviluppo di terapie.
La classificazione clinica storica descrive quattro fasi:
- Fase 1 (Arresto precoce dello sviluppo): Inizia tra i 6 e i 18 mesi, con un lieve rallentamento dello sviluppo. Si possono osservare minore contatto visivo, ridotto interesse per i giocattoli, ritardi nel sedersi o gattonare, e rallentamento della crescita della testa.
- Fase 2 (Rapida regressione dello sviluppo): Inizia tra 1 e 4 anni, con un esordio rapido o graduale. Si verifica la perdita delle abilità manuali mirate e del linguaggio parlato. Compaiono movimenti caratteristici delle mani (strizzare, battere, sfregare, portare alla bocca), spesso assenti durante il sonno. Possono manifestarsi irregolarità respiratorie, instabilità nell'andatura e, in alcuni casi, sintomi simili a quelli del disturbo dello spettro autistico, come compromissione dell'interazione sociale e della comunicazione.
- Fase 3 (Stadio pseudostazionario): Inizia tra i 2 e i 10 anni e può durare diversi anni. Sono comuni convulsioni, deficit motori e aprassia. Talvolta, sintomi come pianto, irritabilità e similitudini con l'autismo tendono a diminuire, mentre aumentano l'allerta, la capacità di comunicazione, l'attenzione e l'interesse per l'ambiente circostante.
- Fase 4 (Deterioramento motorio tardivo): Può durare anni o decenni. Caratteristiche comuni includono scoliosi, ridotta mobilità, debolezza muscolare, spasticità o rigidità. La capacità di camminare può essere persa. La comunicazione visiva diventa prominente in assenza di linguaggio parlato, e i movimenti ripetitivi delle mani possono diminuire. Sono frequenti anomalie cardiache, come il prolungamento dell'intervallo QT. Il rallentamento della crescita e la difficoltà nel mantenimento del peso sono comuni.
La diagnosi della sindrome di Rett è clinica, basata sull'osservazione dei sintomi e dei segni durante le prime fasi di crescita e sviluppo, integrata da valutazioni continue dello stato fisico e neurologico. I criteri diagnostici rivisti da Neul e colleghi (2010) distinguono tra forme tipiche e atipiche. La conferma genetica tramite test per la mutazione del gene MECP2 è fondamentale, ma non sostituisce la valutazione clinica.
I criteri diagnostici richiesti per una diagnosi di sindrome di Rett tipica includono un periodo di regressione seguito da recupero o stabilizzazione, tutti i criteri principali e l'assenza di criteri di esclusione. I criteri principali comprendono la perdita di abilità manuali e del linguaggio parlato, movimenti ripetitivi delle mani e anomalie nella deambulazione. I criteri di esclusione riguardano la presenza di altre patologie con sintomi simili.
I criteri di supporto, pur non essendo strettamente necessari per una diagnosi di forma tipica, possono essere presenti e aiutare nella diagnosi di forme atipiche. Questi includono disturbi respiratori, scoliosi, bruxismo, anomalie del sonno, mani e piedi freddi, disturbi vasomotori, ritardo di crescita, tono muscolare anormale, comunicazione oculare intensa, risate o urla inappropriate e ridotta risposta al dolore.
Disturbo dello Spettro Autistico: Caratteristiche e Diagnosi
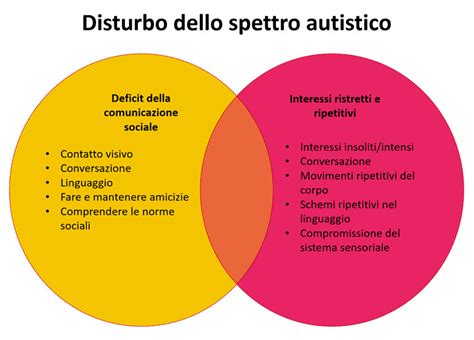
I Disturbi dello Spettro Autistico (ASD) sono classificati tra i disturbi del neurosviluppo e si caratterizzano per un'alterazione delle abilità di socializzazione e comunicazione, che porta a un profilo di sviluppo atipico e variabile nel tempo. Secondo il DSM-5, la diagnosi di ASD richiede la coesistenza di deficit socio-comunicativi con pattern di interessi, attività o comportamenti ristretti o ripetitivi.
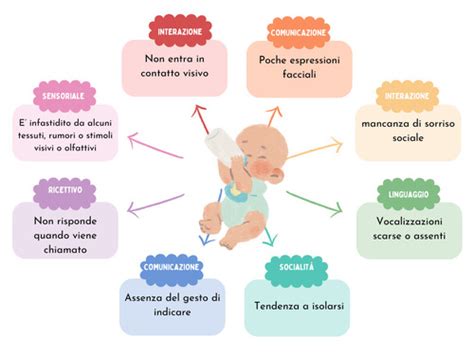
Già dal primo anno di vita, prima ancora dell'emergere della comunicazione verbale, i bambini con ASD possono manifestare segnali di atipie nell'interazione sociale. Questi includono un aggancio oculare sfuggente, la mancanza di comportamenti anticipatori (come tendere le braccia al genitore), difficoltà nella condivisione di piacere o interesse tramite l'uso integrato dello sguardo e dei gesti (come l'indicare, fondamentale per l'attenzione congiunta), e una mancata risposta al proprio nome. Nel gruppo dei pari, si osserva una tendenza all'isolamento o difficoltà nel mantenere relazioni adeguate, nonché una comprensione limitata delle dinamiche intersoggettive e sociali. Nel gioco, le abilità simboliche ("far finta") possono essere assenti o inadeguate all'età. Si aggiungono comportamenti ripetitivi (stereotipie motorie, nell'uso degli oggetti) e/o interessi ristretti e atipici per intensità o età, e rigidità comportamentale.
La diagnosi dei disturbi dello spettro autistico richiede un'attenta valutazione anamnestica, del livello di sviluppo e del funzionamento adattivo. È fondamentale la somministrazione di test diagnostici specifici, interviste semi-strutturate ai genitori e osservazioni del bambino in contesti di gioco. L'impostazione del trattamento beneficia enormemente dall'intervento di un'équipe multidisciplinare composta da neuropsichiatra infantile, psicologo, logopedista e terapista della neuropsicomotricità dell'età evolutiva.
La Distinzione tra Sindrome di Rett e ASD: Sovrapposizioni e Differenze Chiave
Sebbene sia la sindrome di Rett sia i disturbi dello spettro autistico possano presentare deficit nella comunicazione sociale e comportamenti ripetitivi, è cruciale sottolineare le loro differenze fondamentali. La sindrome di Rett è una specifica condizione genetica con un decorso evolutivo caratteristico, che include una fase di regressione motoria e cognitiva dopo un periodo iniziale di sviluppo normale. I disturbi dello spettro autistico, invece, rappresentano un gruppo eterogeneo di condizioni caratterizzate da deficit persistenti nella comunicazione sociale e interazione sociale e da pattern di comportamenti, interessi o attività ristretti e ripetitivi, senza una causa genetica unica e definita come nella RTT.
Il DSM-5-TR (2022) chiarisce che i criteri per la diagnosi di ASD devono essere soddisfatti integralmente e introduce lo specificatore "associato a condizione medica/genetica nota", che può includere la sindrome di Rett. Questo significa che una bambina con sindrome di Rett potrebbe anche soddisfare i criteri diagnostici per l'ASD, ma la diagnosi primaria rimane la sindrome di Rett, data la sua eziologia genetica specifica.
Prospettive Terapeutiche e di Ricerca
Attualmente, non esiste una cura definitiva né per la sindrome di Rett né per i disturbi dello spettro autistico. Tuttavia, la ricerca sta progredendo rapidamente, aprendo nuove prospettive terapeutiche.
Nel contesto della sindrome di Rett, il trattamento si basa su un approccio multidisciplinare focalizzato sulla gestione dei sintomi. Questo include terapia occupazionale, fisica e della comunicazione, programmi educativi speciali, supporto nutrizionale e gestione di comorbilità come epilessia, scoliosi e disturbi respiratori.
Un importante passo avanti è stato l'approvazione da parte della FDA (USA) nel marzo 2023 del trofinetide (DAYBUE), un analogo del frammento N-terminale dell'IGF-1, come primo trattamento per la sindrome di Rett in adulti e bambini di età pari o superiore a 2 anni. Studi clinici, come il trial di fase 3 LAVENDER, hanno dimostrato un modesto miglioramento nell'uso delle mani, nell'andatura e nella comunicazione verbale, sebbene la diarrea e il calo ponderale siano tra gli eventi avversi più frequenti. La presentazione della domanda di autorizzazione all'immissione in commercio in Europa da parte di Acadia è in corso.
Sono inoltre in fase di studio terapie geniche (come TSHA-102 e NGN-401), che mirano a correggere la disfunzione del gene MECP2. I primi dati clinici su coorti pediatriche e adolescenti/adulte mostrano segnali di efficacia, ma rimangono aperte questioni sulla sicurezza a lungo termine, il dosaggio e il controllo dell'espressione genica. Altri approcci terapeutici esplorati includono la modulazione sinaptica, il metabolismo energetico e programmi combinati di riabilitazione e tecnologie assistive.
Per quanto riguarda i disturbi dello spettro autistico, la ricerca si concentra sullo sviluppo di interventi precoci basati sull'evidenza, terapie comportamentali, interventi educativi personalizzati e l'esplorazione di potenziali trattamenti farmacologici per sintomi specifici come l'irritabilità o l'iperattività. L'Istituto Superiore di Sanità sta elaborando linee guida basate sull'evoluzione delle conoscenze fisiopatologiche e terapeutiche, utilizzando la metodologia GRADE.

La comprensione in continua evoluzione della sindrome di Rett e dei disturbi dello spettro autistico, guidata dai progressi nella diagnostica genetica e nella ricerca clinica, offre speranza per un futuro con migliori strategie di gestione e potenziali trattamenti trasformativi per le persone colpite da queste complesse condizioni neurologiche. La collaborazione tra ricercatori, clinici e famiglie rimane fondamentale per affrontare le sfide ancora esistenti e migliorare la qualità della vita di coloro che vivono con queste sindromi.
tags: #codice #autismo #infantile #rett