Il Test di Bayley, nella sua terza edizione (Bayley-III), rappresenta uno strumento fondamentale per la valutazione dello sviluppo nei bambini, coprendo un arco temporale che va da 1 mese a 42 mesi di età. Questo test non è un mero indicatore di intelligenza, ma una complessa batteria di valutazioni che indaga aree cruciali come lo sviluppo cognitivo, del linguaggio, motorio, socio-emotivo e comportamentale adattivo. L'analisi dei risultati del Test di Bayley, soprattutto in contesti clinici che sospettano o indagano disturbi neurologici, offre uno sguardo approfondito sulle potenziali alterazioni dello sviluppo, molte delle quali possono manifestarsi in condizioni ereditarie complesse come le atassie cerebellari.
La Struttura del Test di Bayley-III e la Sua Rilevanza Clinica
La Bayley-III si articola in cinque scale principali, tre delle quali richiedono la somministrazione diretta da parte di un professionista qualificato (scala cognitiva, scala del linguaggio e scala motoria), mentre le restanti due si basano sulle valutazioni dei genitori o caregiver (scala socio-emozionale e scala del comportamento adattivo). Questa struttura consente una valutazione olistica e multidimensionale dello sviluppo del bambino.

La scala cognitiva valuta aspetti quali lo sviluppo senso-motorio, l'esplorazione dell'ambiente, la manipolazione di oggetti, la memoria, la relazione tra concetti e la formazione di idee. La scala del linguaggio si suddivide ulteriormente in comunicazione recettiva (comprensione verbale, vocabolario, comportamenti preverbali) e comunicazione espressiva (sviluppo del vocabolario e della morfosintassi). La scala motoria indaga la motricità fine (manipolazione, presa, risposta tattile) e la motricità globale (postura, equilibrio, movimento dinamico, pianificazione motoria). Le scale socio-emozionale e del comportamento adattivo forniscono informazioni preziose sul modo in cui il bambino interagisce con l'ambiente, gestisce le proprie emozioni e svolge le attività quotidiane.
Il punteggio finale di ogni scala viene espresso utilizzando parametri simili a quelli delle scale di intelligenza, con una media di 100 e una deviazione standard di 16. Prestazioni comprese tra 84 e 116 sono considerate nella norma. Tuttavia, l'obiettivo primario dei test di sviluppo nella prima infanzia non è la determinazione di un quoziente intellettivo, bensì la valutazione del raggiungimento delle tappe fondamentali della crescita e l'identificazione precoce di eventuali ritardi. A differenza dei test di intelligenza, i test di sviluppo come il Bayley-III non assumono che una misura dell'abilità in un dato momento predica necessariamente quella in un tempo successivo, poiché le strutture sottostanti cambiano qualitativamente con la crescita.
Le Atassie Cerebellari: Un Panorama dei Disturbi Neurologici
L'analisi dei risultati del Test di Bayley può rivelare pattern che suggeriscono la presenza di disturbi neurologici complessi, tra cui le atassie cerebellari. Queste condizioni sono caratterizzate da alterazioni della velocità, dell'ampiezza e della forza dei movimenti, principalmente a causa di un malfunzionamento del cervelletto, la struttura cerebrale deputata al mantenimento dell'equilibrio e dei movimenti coordinati occhio-testa-collo.
Il cervelletto, anatomicamente diviso in tre parti (vermis o linea mediana, emisferi cerebellari e lobo flocculo-nodulare), presenta una stretta connessione con i nuclei vestibolari, essenziali per l'equilibrio. Un danno a questo livello comporta alterazioni della postura e dell'andatura, spesso descritte come marcia atassica.
Le atassie possono essere classificate in diverse categorie, tra cui le atassie cerebellari ereditarie, che rappresentano una vasta e complessa famiglia di disturbi neurodegenerativi. Queste sono ulteriormente suddivise in autosomiche dominanti (ADCA) e autosomiche recessive (ARCA).
Atassie Cerebellari Ereditarie Autosomiche Dominanti (ADCA)
Le ADCA, note anche come atassie spinocerebellari (SCA), sono un gruppo eterogeneo di malattie caratterizzate dalla degenerazione progressiva del cervelletto, del tronco encefalico e del midollo spinale. La loro insorgenza varia, ma spesso si manifestano nell'età media giovanile o adulta. Tra le oltre 30 forme identificate, alcune includono:
- SCA1, SCA2, SCA3 (Malattia di Machado-Joseph), SCA6, SCA7, SCA8, SCA10, SCA17, SCA26, SCA30, SCA31: Queste forme sono spesso associate a degenerazioni ereditarie e possono coinvolgere diverse aree del sistema nervoso centrale. La causa genetica è solitamente una espansione di ripetizioni di trinucleotidi (come CAG) in specifici geni. Ad esempio, la SCA6 è causata da una mutazione nel gene CACNA1A, che codifica per un canale del calcio voltaggio-dipendente localizzato nel cromosoma 19p, altamente espresso nel cervelletto. Individui con SCA6 hanno tipicamente tra 20 e 33 ripetizioni della tripletta CAG, e questo tipo di atassia è spesso distinguibile dalla SCA6 classica per la sua risposta all'acetazolamide, sebbene la risposta possa variare.
Le manifestazioni cliniche delle ADCA sono varie e possono includere:
- Atassia della marcia e degli arti: Difficoltà nel camminare, instabilità, goffaggine nei movimenti.
- Dismetria: Difficoltà nel giudicare le distanze e la forza necessaria per un movimento, portando a sfiorare o superare l'obiettivo.
- Disartria: Difficoltà nell'articolazione del linguaggio, che può apparire scansione, lenta o con accento irregolare.
- Tremore intenzionale: Tremore che si manifesta durante il movimento volontario, peggiorando avvicinandosi all'obiettivo.
- Nistagmo: Movimenti oculari involontari e ritmici, che possono essere orizzontali, verticali o rotatori. Possono esserci anche dismetria oculare e scarsa capacità di seguire gli oggetti in movimento.
- Deterioramento cognitivo: In alcune forme, le capacità mentali possono diminuire progressivamente.
- Altri sintomi: Possono includere disturbi del sonno, neuropatia periferica, scoliosi e cardiomiopatia evolutiva.
La diagnosi di queste forme spesso richiede un'attenta valutazione clinica, studi di neuroimaging (risonanza magnetica cerebrale, preferibilmente con sequenze T1, T2, diffusione e FLAIR, per visualizzare il cervelletto e/o il tronco cerebrale) e test genetici per identificare le specifiche mutazioni.
Atassie Cerebellari Ereditarie Autosomiche Recessive (ARCA)
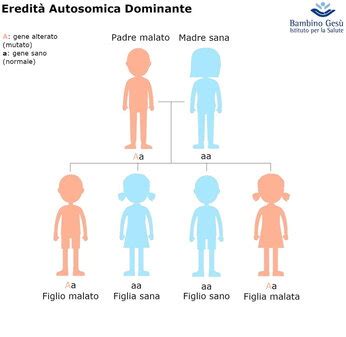
Le ARCA sono un gruppo di disturbi genetici in cui un individuo eredita due copie di un gene mutato (una da ciascun genitore) per sviluppare la malattia. Questo gruppo include alcune delle atassie più note e studiate.
Atassia di Friedreich (FRDA)
L'Atassia di Friedreich è la forma più comune di atassia ereditaria autosomica recessiva, con una prevalenza stimata tra 1:29.000 e 1:50.000 nati vivi. La causa è una mutazione nel gene FXN, situato sul cromosoma 9, che codifica per la proteina fratassina. La fratassina è cruciale per l'omeostasi del ferro nei mitocondri, e il suo deficit porta a un accumulo di ferro, stress ossidativo e danno alle proteine, in particolare quelle contenenti centri ferro-zolfo.
La mutazione più comune è un'espansione di ripetizioni di triplette GAA all'interno del primo introne del gene FXN, che porta a una ridotta espressione della fratassina. La gravità del fenotipo è correlata all'estensione della ripetizione GAA e alla presenza di mutazioni puntiformi.
Le caratteristiche cliniche dell'Atassia di Friedreich includono:
- Atassia progressiva: Inizia tipicamente con goffaggine nella marcia, difficoltà nella stazione eretta e nella corsa, evolvendo verso una marcia tabetocerebellare. L'atassia colpisce tronco, gambe e braccia.
- Debolezza muscolare: Progressiva debolezza degli arti inferiori, con riflessi osteotendinei aboliti o ridotti.
- Disfunzioni sensoriali: Neuropatia assonale sensitiva, che può manifestarsi con perdita di propriocezione e sensibilità vibratoria.
- Disfunzioni cardiache: La cardiomiopatia ipertrofica è una complicanza frequente e la causa più comune di morte. Anomalie dell'onda T e segni di ipertrofia ventricolare sono comuni all'ECG.
- Deformità scheletriche: Piede cavo, piede equinovaro, dita ad artiglio e scoliosi sono frequenti.
- Disfagia e disartria: Difficoltà nella deglutizione e nell'articolazione del linguaggio compaiono nelle fasi più avanzate.
- Perdita d'udito neurosensoriale: Osservata in una percentuale di pazienti.
- Diabete mellito: Può verificarsi in alcuni individui.
L'esordio dei sintomi avviene mediamente intorno ai 15.5 anni, ma può variare da 2-3 anni a oltre 25 anni. La progressione è variabile, ma molti pazienti necessitano di una sedia a rotelle entro la fine della quarta decade di vita. La diagnosi si basa sulla clinica e viene confermata da test genetici che rilevano l'espansione delle triplette GAA nel gene FXN.
Il percorso riabilitativo sull’Atassia di Friedreich presso La Nostra Famiglia di Conegliano Veneto
Atassia-Telangiectasia (AT)
L'Atassia-Telangiectasia, nota anche come Sindrome di Louis-Bar, è una malattia genetica autosomica recessiva rara che colpisce circa 1 su 100.000 nati vivi. È causata da mutazioni nel gene ATM, situato sul cromosoma 11q22-23, che codifica per una proteina chinasi fondamentale nella risposta al danno del DNA, in particolare alle rotture del doppio filamento (DSB).
Le caratteristiche distintive dell'AT includono:
- Atassia cerebellare: Si manifesta nei primi anni di vita, con alterazioni della postura e dell'andatura. I bambini possono ruotare la testa anziché muovere gli occhi per seguire un oggetto.
- Telangiectasie: Dilatazioni dei piccoli vasi sanguigni, visibili principalmente negli occhi (congiuntiva) e sulla pelle (soprattutto nelle pieghe del gomito e del ginocchio).
- Immunodeficienza: Deficit immunitari, in particolare nel sistema immunitario umorale e cellulare, che rendono i pazienti suscettibili a infezioni ricorrenti, specialmente respiratorie. Possono sviluppare bronchiectasie.
- Aumentato rischio di tumori: I pazienti con AT hanno un rischio significativamente aumentato di sviluppare tumori, in particolare leucemie e linfomi, a causa della loro ridotta capacità di riparare il danno al DNA e della loro radiosensibilità.
- Sensibilità alla radioterapia e ad alcune chemioterapie: A causa della ridotta capacità di riparazione del DNA, questi pazienti sono particolarmente vulnerabili agli effetti dannosi delle radiazioni ionizzanti e di alcuni farmaci chemioterapici, che devono essere usati con estrema cautela.
- Altre manifestazioni: Possono includere disfunzioni endocrine, ritardo della crescita e, in alcuni casi, diabete.
La diagnosi viene confermata da test genetici che identificano le mutazioni nel gene ATM e da studi che rilevano la radiosensibilità e l'instabilità cromosomica (ad esempio, traslocazioni 7;14). Molti pazienti affetti necessitano di una sedia a rotelle all'età di 10-11 anni e la sopravvivenza media è di circa 25 anni, anche se forme con caratteristiche più miti e sopravvivenza più lunga sono state descritte.
Altre Atassie Cerebellari Autosomiche Recessive
- Atassia-teleangiectasia oculomotoria (AT-OM): Una variante dell'AT con caratteristiche simili, ma con una maggiore enfasi sui disturbi oculomotori.
- Degenerazioni cerebellari ereditarie o tossine: Oltre alle forme genetiche, è importante considerare cause acquisite o tossiche che possono portare a degenerazione cerebellare.
Disturbi Correlati e Diagnosi Differenziale
È fondamentale distinguere le atassie cerebellari da altre condizioni neurologiche che possono presentare sintomi simili.
Sindrome dell'X Fragile (FXS)
La Sindrome dell'X Fragile è la causa genetica più comune di disabilità intellettiva ereditaria dopo la sindrome di Down. È causata da una mutazione nel gene FMR1 sul cromosoma X, che comporta un'espansione di ripetizioni della tripletta CGG. Questo porta alla metilazione del gene e alla soppressione della produzione della proteina FMRP (Fragile X Mental Retardation Protein), essenziale per lo sviluppo neuronale.
Le caratteristiche della FXS includono:
- Disabilità intellettiva: Di gravità variabile, da lieve a grave.
- Caratteristiche facciali: Volto allungato, fronte prominente, orecchie grandi, macroorchidismo (testicoli ingrossati) negli uomini.
- Problemi comportamentali: Iperattività, deficit di attenzione, autismo o tratti autistici.
- Problemi motori: A volte presenti, come tremori e atassia, sebbene non siano il sintomo principale.
Nei portatori della premutazione del gene FMR1, possono svilupparsi condizioni come la FXTAS (Fragile X-Associated Tremor/Ataxia Syndrome), che si manifesta con tremore, atassia e deterioramento cognitivo in età adulta, e la FXPOI (Fragile X-Associated Primary Ovarian Insufficiency), caratterizzata da insufficienza ovarica prematura. La diagnosi della FXS richiede test genetici specifici per la mutazione del gene FMR1.
Malattia di Von Hippel-Lindau (VHL)
La Malattia di Von Hippel-Lindau è una sindrome ereditaria autosomica dominante caratterizzata dalla predisposizione allo sviluppo di tumori in vari organi. È causata da mutazioni nel gene VHL, un gene oncosoppressore.
Le manifestazioni più comuni includono:
- Emangioblastomi: Tumori benigni ma potenzialmente pericolosi che si sviluppano frequentemente nel cervelletto, nel tronco encefalico e nel midollo spinale. Questi tumori possono causare sintomi neurologici come mal di testa, nausea, vomito, deficit motori e atassia, a causa della compressione delle strutture nervose.
- Carcinomi a cellule renali.
- Feocromocitomi: Tumori delle ghiandole surrenali.
- Cisti pancreatiche e tumori neuroendocrini del pancreas.
La diagnosi si basa sulla storia familiare, sull'esame clinico e su test genetici per il gene VHL. Il monitoraggio regolare è essenziale per la diagnosi precoce e la gestione dei tumori.
Altre Condizioni Cerebellari
- Malformazione di Chiari: Anomalie congenite in cui il tessuto cerebellare si estende nel canale spinale.
- Tumori del cervelletto: Come medulloblastomi e astrocitomi cistici, che possono causare sintomi cerebellari e, in caso di aumento della pressione endocranica, edema della papilla.
- Infezioni virali: Alcune infezioni virali possono causare atassia cerebellare acuta o subacuta, spesso reversibile.
L'Importanza della Valutazione Multidisciplinare e della Ricerca
La diagnosi e la gestione dei disturbi neurologici, in particolare delle atassie cerebellari, richiedono un approccio multidisciplinare che coinvolga neurologi, genetisti, neuroradiologi, terapisti fisici e occupazionali, logopedisti e psicologi. La Bayley Scales of Infant and Toddler Development - Third Edition (BAYLEY-III) è uno strumento prezioso per identificare precocemente i bambini a rischio di ritardi dello sviluppo, fornendo indicazioni fondamentali per la pianificazione di interventi riabilitativi mirati.
La ricerca continua a svolgere un ruolo cruciale nella comprensione di queste complesse patologie. L'identificazione di nuovi geni associati alle atassie, lo studio dei meccanismi molecolari alla base della neurodegenerazione e lo sviluppo di nuove strategie terapeutiche, inclusa la terapia genica e farmacologica, offrono speranza per migliorare la qualità della vita dei pazienti e delle loro famiglie. La comune pratica clinica si avvale sempre più di test genetici per confermare diagnosi e offrire consulenza genetica, fondamentale per le famiglie affette da queste condizioni ereditarie. La comprensione delle cause non diagnosticate di sintomi cerebellari e l'ampliamento della conoscenza dello spettro fenotipico di malattie come l'Atassia di Friedreich sono aree di ricerca attive.
tags: #bailey #test #autism #risultati #italino